

|
The Vaxign2 Tutorial
Notes:
Introduction: Vaxign2 is a web-based software program in VIOLIN that targets for vaccine design. Based on the reverse vaccinology strategy, Vaxign2 predicts vaccine targets by bioinformatics analysis of genome sequences. Genome sequences come from pathogenic strains, non-pathogenic strains (optional), and host species (human, mouse, or pig). Predicted features in the Vaxign2 pipeline include protein subcellular location, transmembrane helices, adhesin probability, conservation among pathogenic strains, sequence exclusion from genomes of non-pathogenic strains, sequence similarity to host proteins, and epitope binding to MHC class I and class II. Vaxign2 contains precomputed predictions for over 398 genomes and also allows dynamic vaccine target prediction based on users’ input sequences. A video tutorial is available as an alternative or supplement to the written tutorial. The following tutorial will provide step-by-step instructions on how to navigate through the Vaxign2 website (https://violinet.org/vaxign2). Two use cases will be tested: (1) Human SARS coronavirus 2 (SARS-CoV-2) isolate Wuhan-Hu-1 vaccine target prediction; (2) Enterohemorrhagic Escherichia coli (EHEC) O157:H7 vaccine target prediction. The SARS-CoV-2 use case will primarily demonstrate Vaxign2 sequence conservation analyses and MHC class I epitope prediction techniques. The O157:H7 use case will primarily demonstrate general features for bacterial vaccine target prediction including prediction of secreted and outer membrane proteins and transmembrane helices. Both tutorials will use pre-computed results to demonstrate the feature and save time. This tutorial will also demonstrate how to set up your own VIOLIN account to save query results and facilitate collaborative analysis with others.
First use case: SARS-CoV-2 Vaccine Target Prediction Visit the Vaxign2 job submission page. Visit https://vWiolinet.org/vaxign2 in a modern web browser of your choice. Vaxign2 is freely available for public use without any requirement to sign a license or create an account; Creating a VIOLIN account to save your query results for later personal or collaborative use is completely optional. The Vaxign2 job submission page (Figure 1) allows you to submit Vaxign2 job by uploading or entering your own sequence input for Dynamic Analysis, or by choosing a genome that has been pre-processed using the Precompute Query tab (Figure 2). 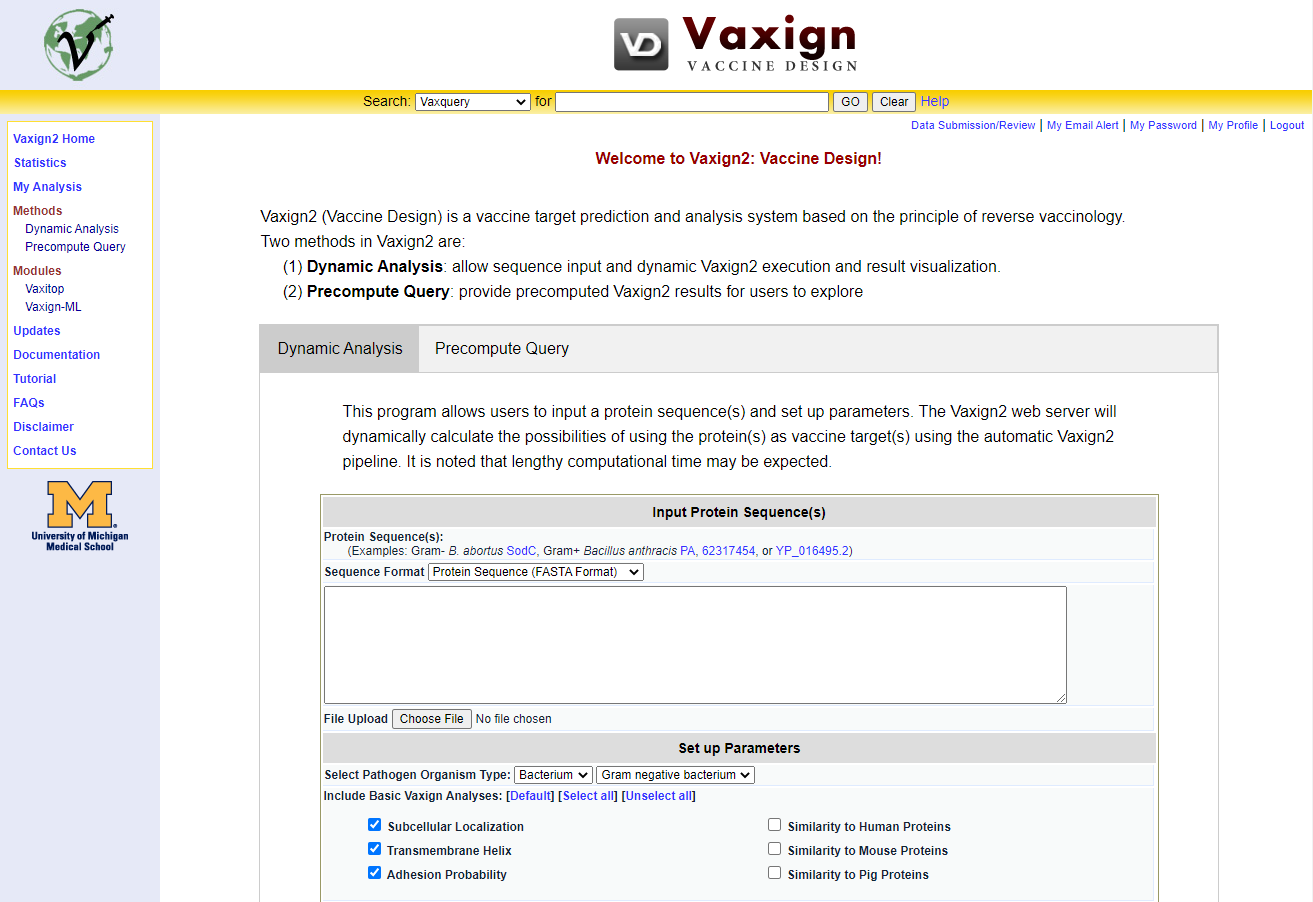
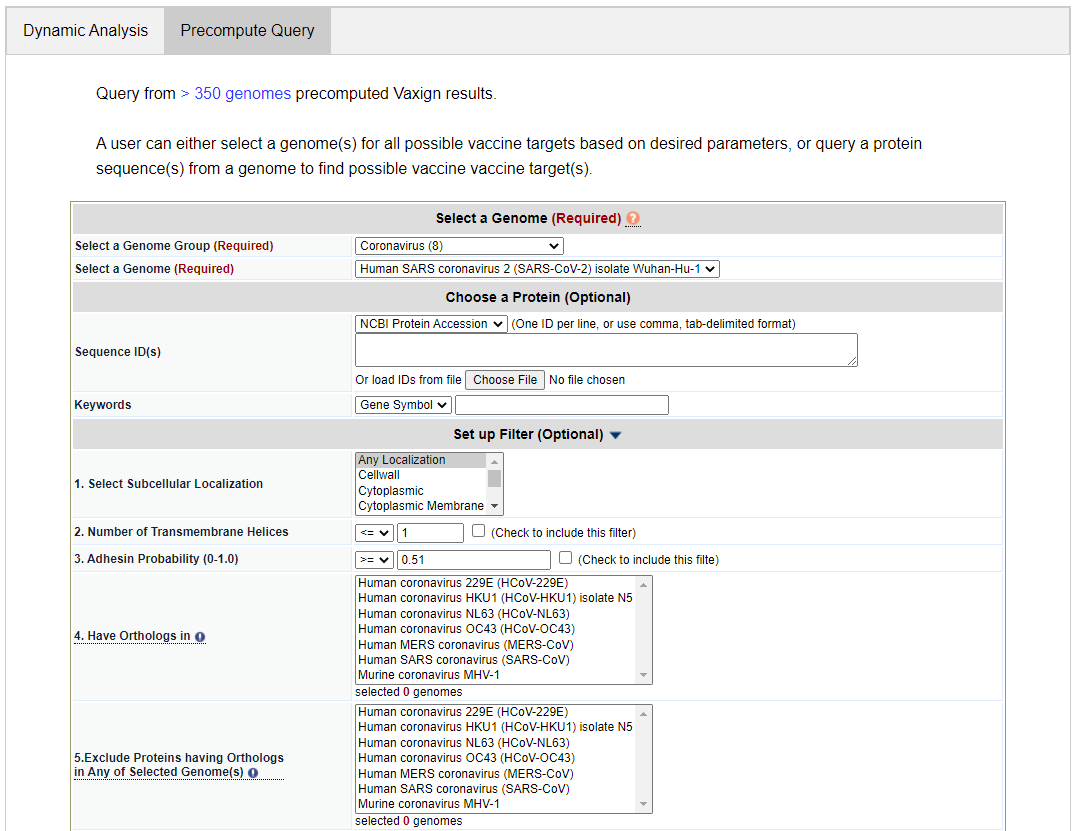
Select a Genome Group. In the Precompute Query tab, click the “Select a Genome Group” dropdown list. Select the option “Coronavirus (8)”. The number “8” means that this group contains 8 genomes, which are from 8 herpesvirus strains. Select a Genome. From the drop down list, select “Human SARS coronavirus 2 (SARS-CoV-2) isolate Wuhan-Hu-1”. Ignore “Sequence ID(s)” This is for querying one or a list of sequence IDs from the specified genome. The IDs can be NCBI Protein Accession number, NCBI Protein GI, NCBI Gene ID, and NCBI Locus Tag. Ignore “Keywords” line. This is for querying one or more proteins based on a specific database ID (e.g., NCBI Locus Tag), gene symbol, or protein description. Select Filter Options This allows you to set criteria to filter out proteins:
For the purposes of the tutorial, do not specify any filter options. Submit your query. Click the “Submit” button in the bottom of the Vaxign2 Query web interface.
Query result examination. After Vaxign2 finishes running the job, you will be redirected to the result page for your query. Thet total number of proteins identified will be displayed above the results table (Figure 3) which displays 50 proteins per page by default. You may change how many entries are shown per page as well as navigate between result pages below the results table. Each row of the table represents a specific protein which includes: (1) Protein Accession number, (2) Protein Name, (3) Gene Accession Number, (4) Gene Symbol, (5) Locus Tag, (6) Vaxign-ML score, (7) Localization, (8) Adhesin Probability, (9) Trans-membrane helices, (10) Similarity to Human Proteins, (11) Similarity to Mouse Proteins, and (12) Similarity to Pig Proteins. 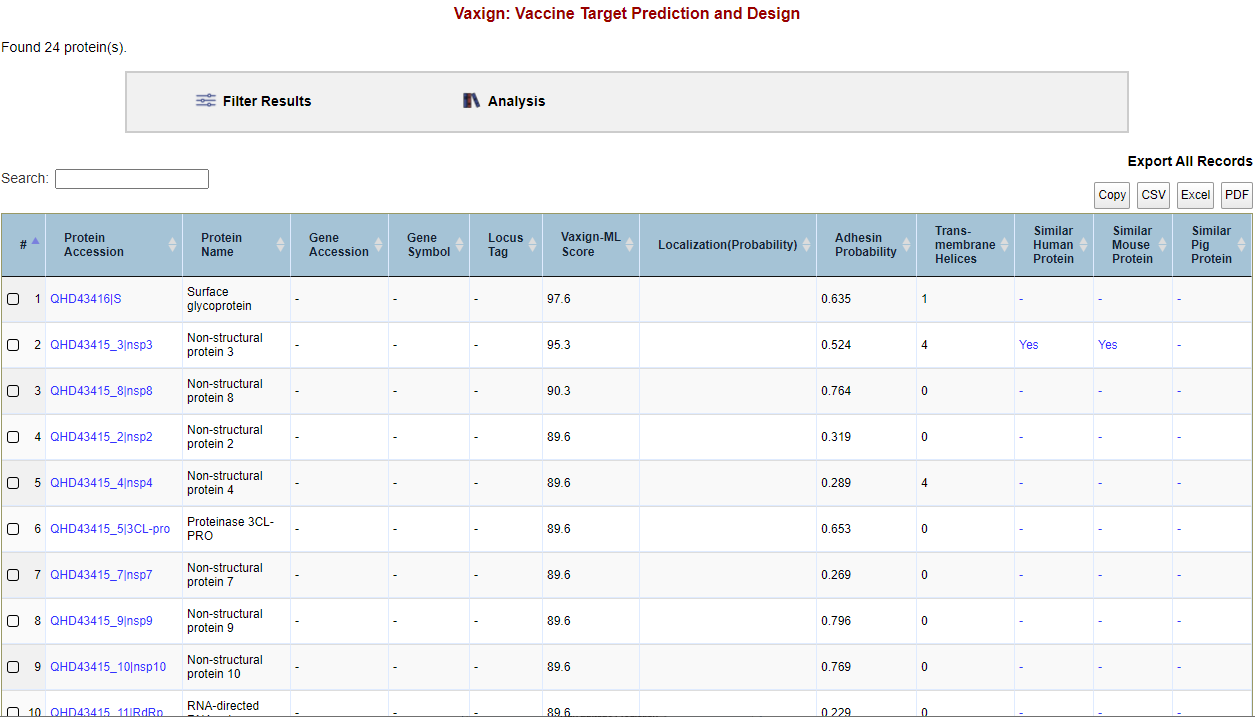
Conservations among human coronaviruses Click “Analysis“ above the results table and then click “Show Genome Group [Coronavirus] Orthologs“. This will take you to a page that will allow you to select other coronaviruses to include in an ortholog table. Select all human coronaviruses, and then click “Submit“. An ortholog table will appear (Figure 4) showing that 10 proteins are conserved between the coronavirus genome group. 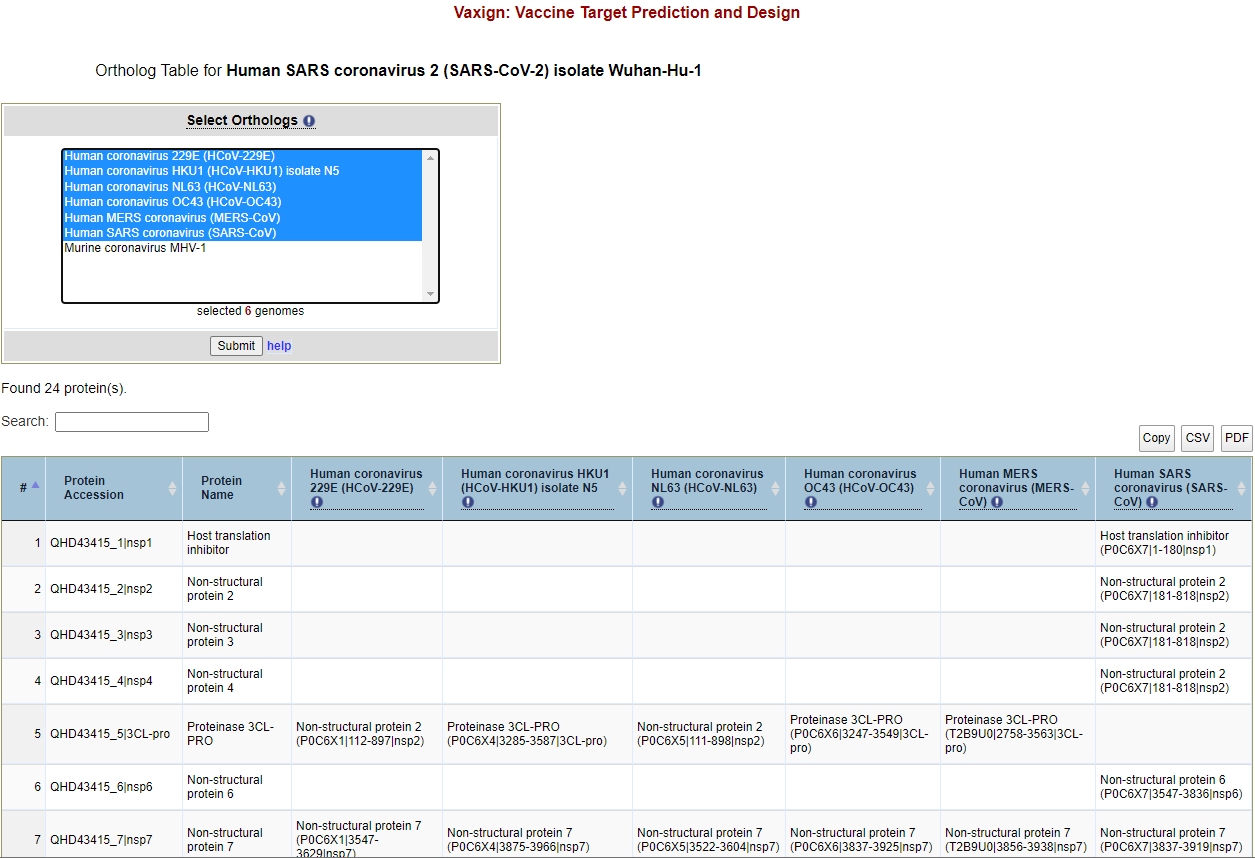
Restriction of maximum number of Transmembrane Helices through filtering Back on the query result page, click “Filter Results“ above the results table. Once the filter frame appears, choose the “less than or equal to" (<=) option in the dropdown field and enter “1“ in the text field to the right of the “Number of Transmembrane Helices“ label. This specifies a view of results for proteins with one or zero transmemebrane helices. To apply the filter, click the checkbox to the right of the quantity field and click “Submit“ at the top of the filter frame. The results will be updated, showing 19 proteins after filtering out 5 proteins that had more than one transmemebrane helix. Other filter rules may be applied in the filter frame. Sort results by Adhesin Probability. By default, Vaxign2 sorts results in descending order by Vaxign-ML Score. To change how the results are sorted, click the “Adhesin Probability“ column header once to sort the results by Adhesin Probability in ascending order, then once more for descending order. Checking detailed results for one protein. The SARS-CoV-2 S protein (NCBI protein ID QHD43416|S) is a commonly used vaccine antigen in current COVID-19 vaccine development. In this example of Vaxign2 predicting a high adhesin probability and Vaxign-ML predicting the same protein as a good vaccine antigen with a score of 97.6, we will investigate the single protein in detail. Click the protein “QHD43416|S“. This will take you to a page with detailed predictions for the protein (Figure 5). From here, you may use the subnavigation to the left of the analysis window to switch between analyses tools. 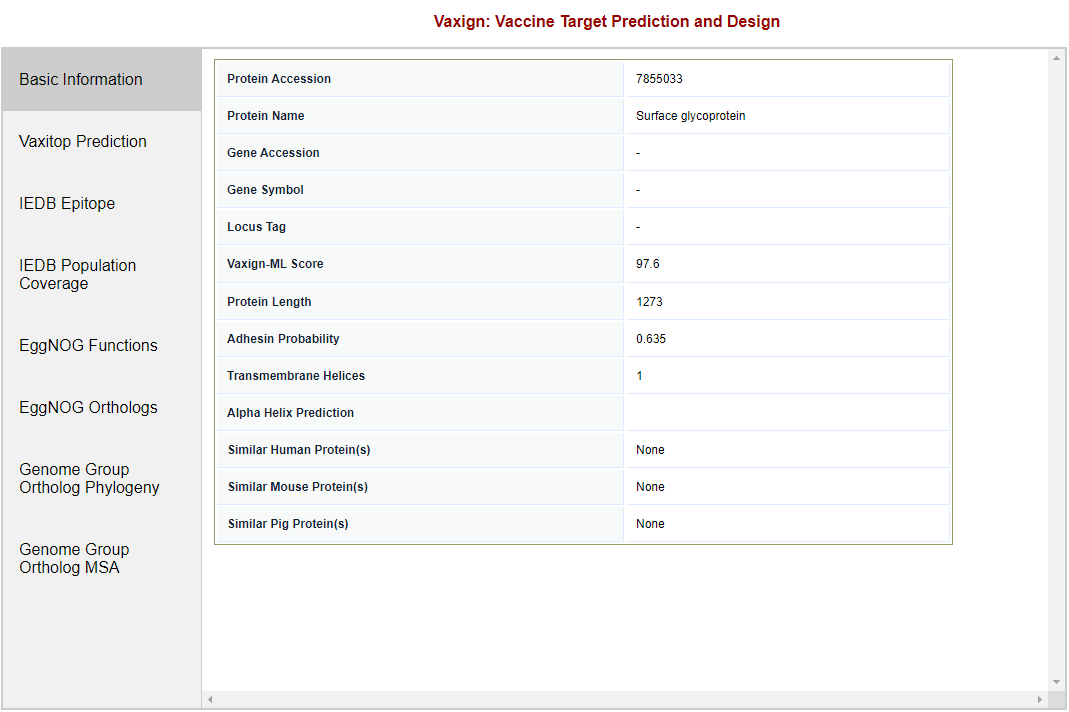
Vaxitop epitope prediction. Alleles may be selected with options for host, MHC class, MHC allele, and epitope length by selecting desired options in their respective dropdown menus and clicking “Add Allele“. Frequently used alleles may be added with a single click in the “Frequently used alleles“ section. Once the desired alleles have been selected, click “Show Vaxitop Results“ to populate the results table. Results may be filtered by p-value cutoff and a general search term for all fields in their respective text fields above the results table. Results may be copied to your clipboard or exported as a CSV, Excel file, or PDF file using the respective buttons directly above the results table. By default, the table will only show 25 entries per results page. You may change this by selecting an alternative number in the dropdown menu below the bottom-left corner of the results table. You may view the next number of results according to your desired display interval by clicking on the the page number links or “Previous“ and “Next“ links to view the results page behind or ahead the page currently being observed. 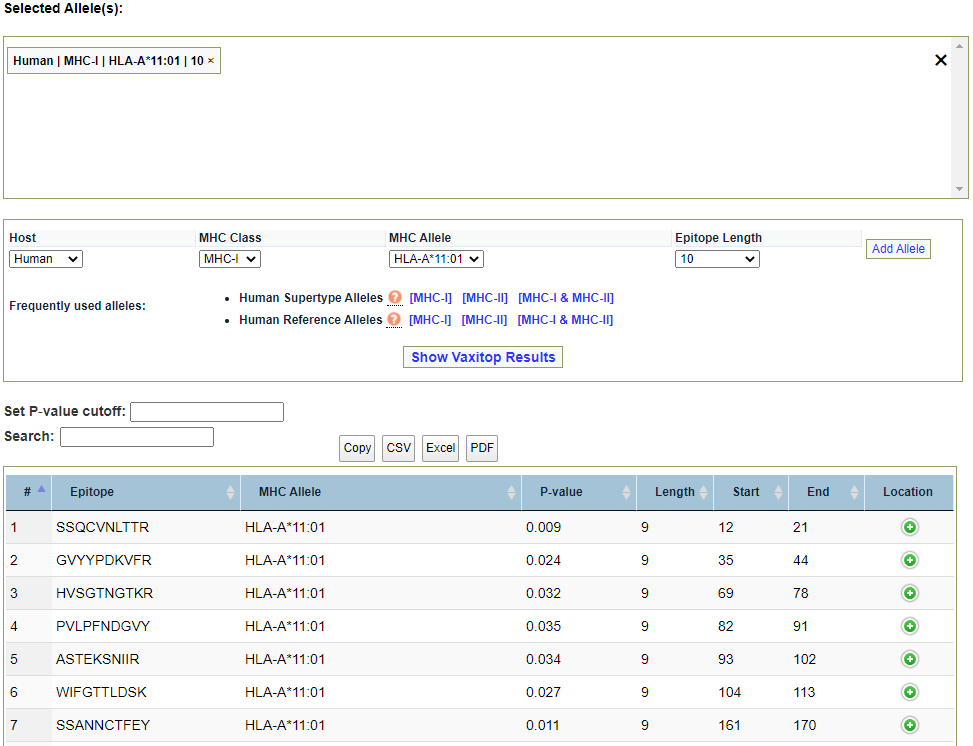
IEDB Epitopes. IEDB experimentally validated epitopes for B Cells or T Cells may be reviewed for a protein of interest. A results table will appear with information including IEDB ID, epitope, starting position, and ending location. Search, export, and results table navigation options behave the same as the Vaxitop prediction screen. 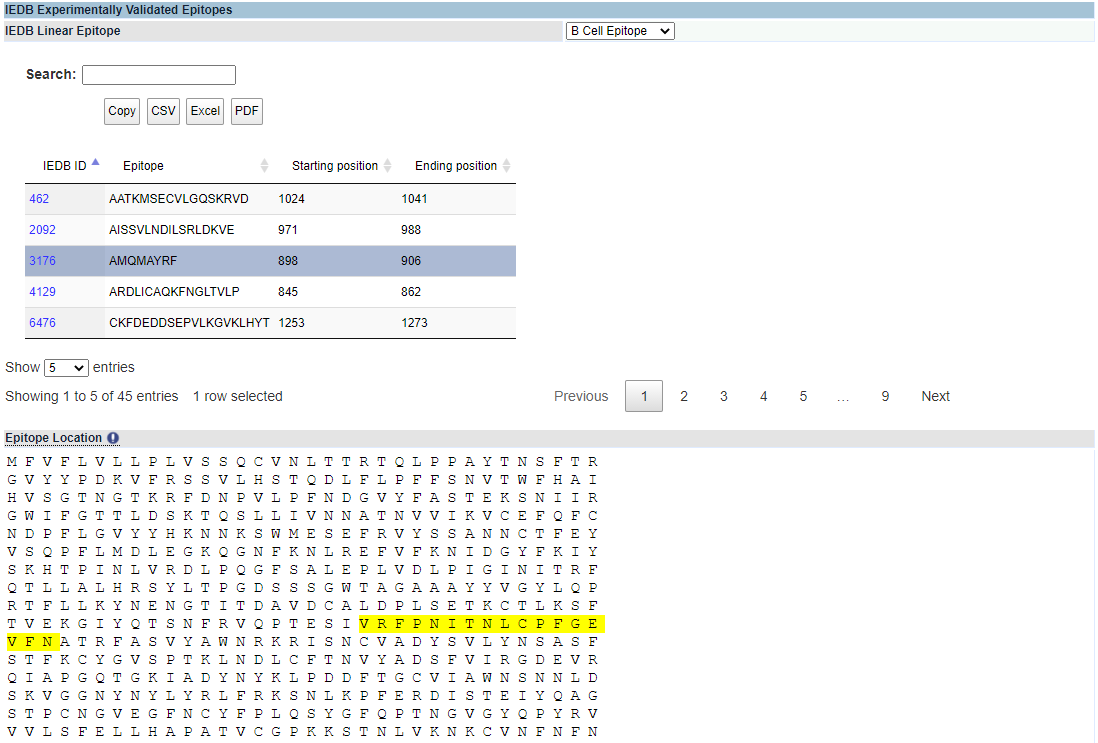
IEDB Population Coverage. Global population coverage for MHC-I, MHC-II, as well as MHC-I and MHC-II combined may be selected in the dropdown menu in the upper left corner of the analysis screen. Map navigational, selection, and image download tools will appear at the top right corner of the map after the cursor is hovered over the map. Countries may be hovered over with the cursor to see coverage corresponding to the selected MHC option. 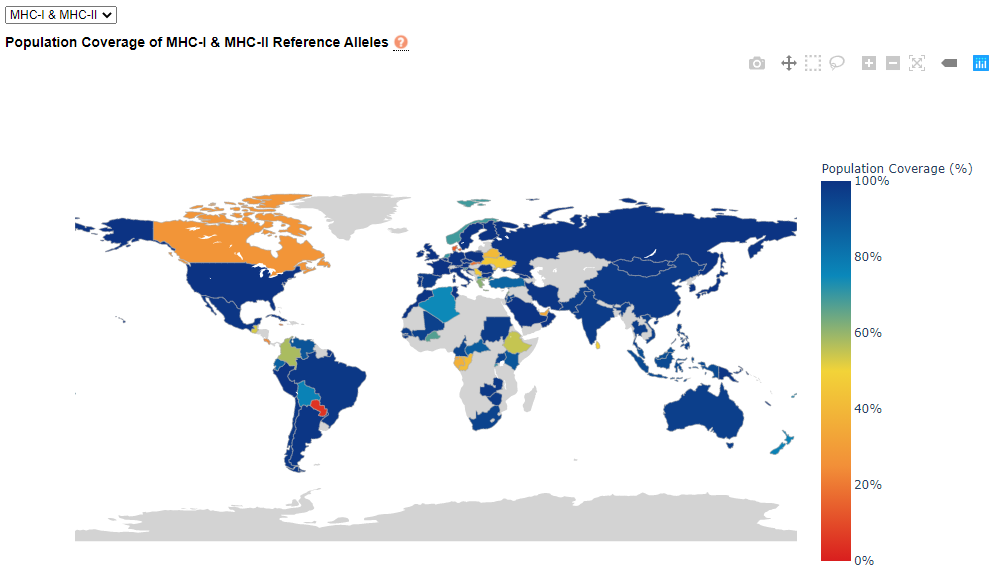
EggNOG Functions. Functional annotations including Clusters of Orthologous Groups (COG) and Gene Ontology (GO) terms may be viewed on this analysis screen. 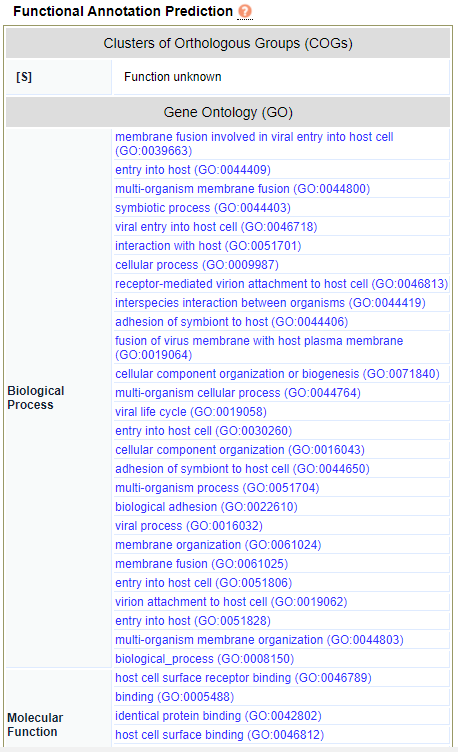
EggNOG Orthologs. This screen displays orthologous proteins predicted by the EggNOG database in a Table View that may be viewed, navigated, and matched against a search term in a similar fashion to previous analysis screens. You may switch from Table View to a taxonomic Tree View by selecting the desired option in the dropdown box to the right of "Orthologous Protein Prediction" 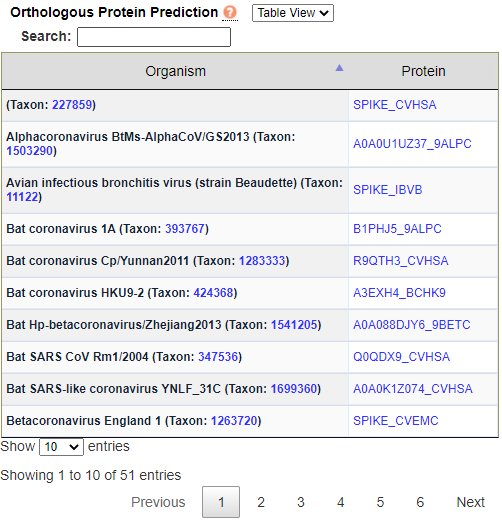

Genome Group Ortholog Phylogeny. This screen displays a phylogenic tree for related organisms and orthologous proteins. A download feature appears in the top-right corner of the analysis screen after hovering the cursor over the tree. This feature is only available to logged in users. 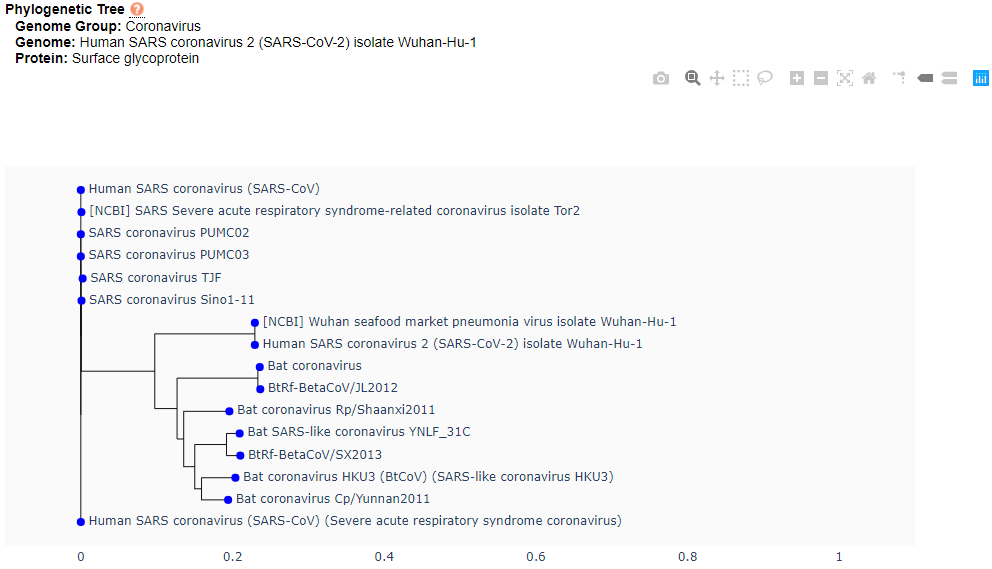
SECOND USE CASE: E. COLI O157:H7 VACCINE TARGET PREDICTION We will not provide much detail for this use case here. The procedure to run this is similar to what we explain above. One feature that is not demonstrated in the above example is the Filter Option:
This filter method is based on PSortb (http://www.psort.org/psortb/), which is specifically designed for prediction of subcellular localization of proteins from Gram + or Gram – bacteria. It is not designed for other types of microbes. Figure 7 provides some concrete settings for an example analysis. In this example, we chose to identify only those extracellular and outer membrane proteins. This analysis results in 45 hits. 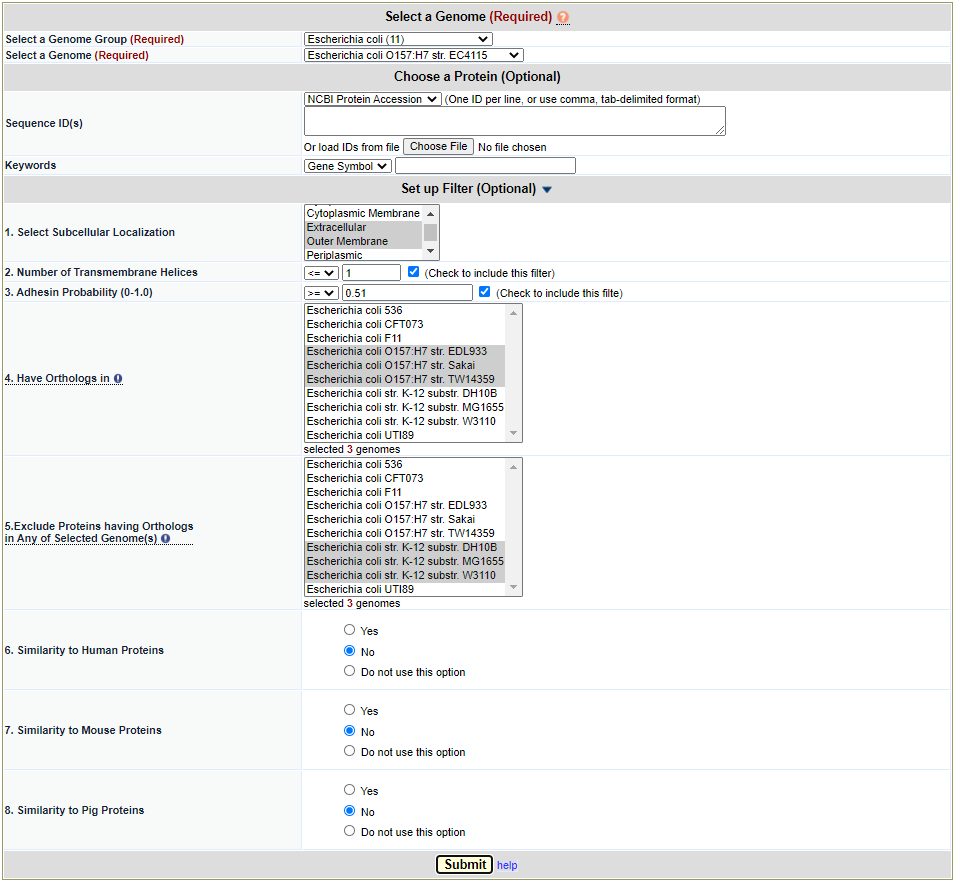
Register and Use a Vaxign2 Account for Predicted Result Storage and Sharing 
Exit Vaxign2. Once you complete your exercises, you may log out of Vaxign2 if you generate your own account and log in. To log out, navigate to the top of the window, and click on LOGOUT.
------------------------ Note: Please see more information in our Vaxign2 Documentation page.
Provide Comments. Once you complete your exercises, you may log out of Vaxign2 if you generate your own account and log in. In an effort to improve the Vaxign2 performance and provide better support for the vaccine development community, we would appreciate any comments/suggestions you may have regarding the Vaxign2 program. To provide your comments, please email us or fill out a feedback online form in the Vaxign2 Contact Us page. Thank you!! (URL: https://violinet.org/vaxign2/vaxitop). Also, although the Vaxitop epitope prediction selection is not available in the cover page, the Vaxitop epitope prediction method is available for any protein you select after the initial screening. To predict MHC class I and II epitopes, the Vaxitop method is based on prediction of position specific scoring matrices (PSSM). Different from existing epitope prediction algorithms, Vaxitop relies on statistical P-value (instead of a percentage or top number) as the cutoff. A P-value of 0.05 provides a cutoff with high and balanced sensitivity and specificity. Under this section, you can choose a P-value cutoff, host species, MHC allele, and epitope length. It’s noted that the Vaxitop |